Fibrosis Quística


"Tú respiras sin pensar... yo sólo pienso en respirar"
Lema de la Federación Española de Fibrosis Quística
CFTR:

Proteína de 1.480 aminoácidos que pertenece a una familia de transportadores llamada ABC (ATP Binding Cassette) que permiten el transporte bidireccional de Cl- y otros aniones a través de las membranas, mediante la unión de ATP y la fosforilación de los dominios reguladores, como podemos ver en la Fig 1. Estos transportadores se localizan en la cara externa de las células y juegan un papel muy importante en la regulación funcional de órganos con tejidos secretores y/o absortivos como pulmón, páncreas, glándulas sudoríparas, etc.
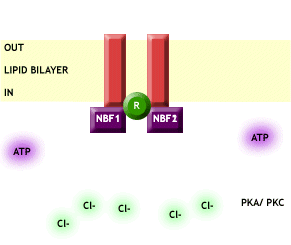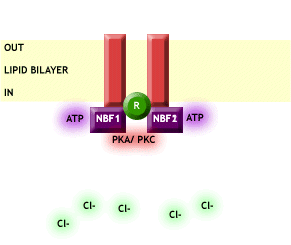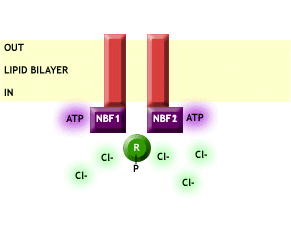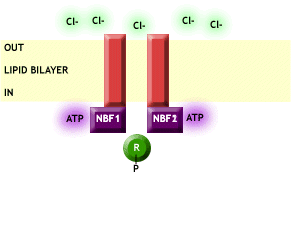
Fig. 1 CFTR. Podemos observar como el canal, mediante la unión de ATP y la fosforilación de los dominios reguladores (R), permite el paso de Cl- a través de la membrana plasmática. Imagen extraída de http://www.genemedresearch.ox.ac.uk/cysticfibrosis.html
Esta proteína no sólo regula el transporte de Cl- sino también es muy importante su función reguladora sobre un gran número de procesos celulares y de otros transportadores de iones, por ejemplo el de sodio (Na+), como vemos en la Fig. 2. [3]

Fig. 2 El CFTR activo permite el paso de Cl- a través de la membrana plasmática y a su vez regula la entrada de Na+. Imagen extraída de http://www.genemedresearch.ox.ac.uk/cysticfibrosis/function.html
En el momento que CFTR desparece o no es funcional debido a alguna mutación, se pierde el transporte de Cl- a través de la membrana plasmática y a su vez se pierde la regulación que efectúa el CFTR sobre el resto de canales, provocando entre otras cosas una entrada excesiva de Na+ a las células, como vemos en la Fig. 3.
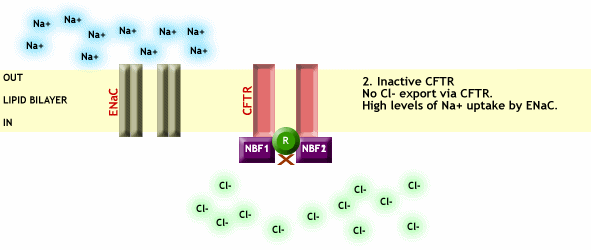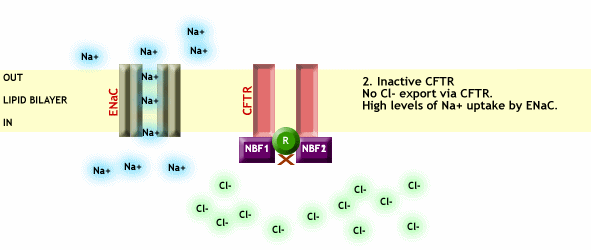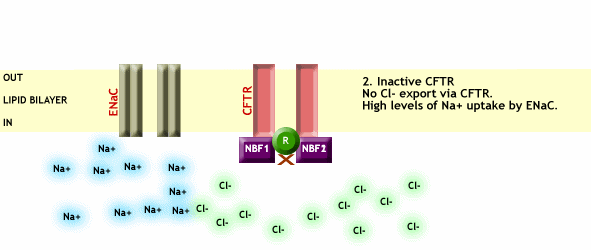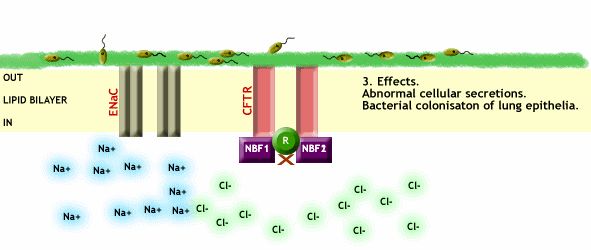
Fig. 3 CFTR no funcional, impide el paso de Cl- a través de la membrana plasmática y a su vez no ejerce ningún tipo de regulación sobre los canales de Na+, provocando así una entrada excesiva de este ión dentro de las células. Imagen extraída de http://www.genemedresearch.ox.ac.uk/cysticfibrosis/function.html
Se han descrito unas 1000 mutaciones diferentes que pueden alterar la función normal del CFTR. Estas mutaciones las podemos clasificar en diferentes tipos según el mecanismo molecular que alteran o en función de las manifestaciones clínicas que provocan. [1, 3]
Clasificación de las mutaciones de la CFTR:
Según el mecanismo molecular alterado
Mutaciones tipo I: biosíntesis de la proteína alterada.
Mutaciones tipo II: maduración de la proteína alterada.
Mutaciones tipo III: afectan a la regulación y obertura del canal.
Mutaciones tipo IV: afectan a la conductancia del canal.
Mutaciones tipo V: afectan a la estabilidad de la proteína.
Mutaciones severas: no hay proteína funcional (I, II, III).
Mutaciones suaves: hay cierta proteína funcional (IV, V).
Según las manifestaciones clínicas

Fig. 4 Mutaciones del CFTR según el mecanismo molecular alterado. Imagen modificada de http://jillclare.tripod.com/cf/
La mutación más común es la pérdida del aminoácido 508 (Phe), mutación conocida como ΔF508, que da lugar a una mutación de tipo II. Esta mutación presenta una incidencia de 1:25 y para poder explicar la alta prevalencia de esta mutación, se han propuesto varias teorías:
- Efecto fundador.
- Ventaja selectiva del heterocigoto a determinadas enfermedades como el cólera.
Ninguna de estas teorías está ampliamente aceptada.
